Androgen Metabolism
Authors
INTRODUCTION
Androgen metabolism, in the broadest sense, includes the secretion, transport, tissue uptake, peripheral transformation, and excretion of C-19 steroids. Increased knowledge of androgen metabolism has widened the concept of the function of steroid metabolism to include more than just inactivation and excretion. Metabolic transformation of secreted androgens produces steroids with variable effects and potency; some, in fact, are estrogens.
Androgen metabolism varies with sex, age, thyroid status, and weight and from tissue to tissue. Androgenicity is not due exclusively to the androgen production rate but is also influenced by the process of metabolism. The interactions of those aspects of androgen metabolism that decrease or enhance androgenic effects are emphasized.
ANDROGEN SYNTHESIS AND SECRETION
The major androgens produced by the adult woman in decreasing order of magnitude are dehydroepiandrosterone sulfate (DHEAS), dehydroepiandrosterone (DHEA), androstenedione (Δ4-A), androstenediol (Δ5-A-diol), testosterone (T), and dihydrotestosterone (DHT).1 The mean plasma concentrations of these androgens in the premenopausal woman are listed in Table 1.
Table 1.Mean Plasma Concentrations of the Major Androgens in Normal Premenopausal Females
| Plasma Concentration | |
Androgen | ng/ml | nmoles/liter |
DHEAS | 1700 | 4630 |
DHEA | 4.2 | 14.6 |
Δ4-A | 1.76 | 6.1 |
Δ5-A diol | 0.75 | 2.6 |
T | 0.39 | 1.3 |
DHT | 0.19 | 0.65 |
DHEAS, dehydroepiandrosterone sulfate; DHEA, dehydroepiandrosterone; Δ4-A, androstenedione; Δ5-A diol, androstenediol; T, testosterone; DHT, dihydrotestosterone.
The main pathways of androgen metabolism and the required enzymes are depicted in Figure 1. The major sites that are involved in metabolism are the C-17, C-3, and C-5 positions of the steroid, and these are the same sites that determine androgenic activity.2 These pathways are present in the adrenals, ovaries, and peripheral tissues. The conversion of DHEA to Δ4-A or Δ5 - A-diol to T requires the 3β-hydroxy-Δ5 - steroid dehydrogenase enzyme systems (3β-HSD/KSI), which convert the unsaturated bond from the C-5,6 position to the C-4,5 position and oxidizes the 3β-hydroxy group to a 3-keto group. Recent studies have shown that there are a number of isoforms of this enzyme with tissue specificity. They belong to the family of short-chain dehydrogenase/reductase family.3 The interconversion of the 17-keto and the 17β-hydroxyl (OH) complex is accomplished by the 17β-hydroxysteroid dehydrogenase enzyme.
|
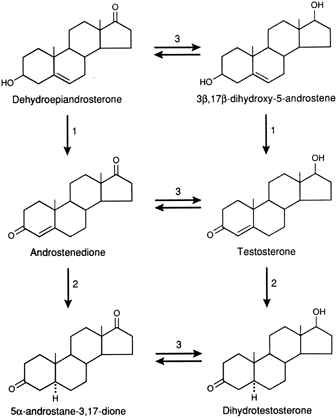
There are at least five isoenzymes that are cell and substrate specific.4 Some of the isoenzymes convert 17-keto steroids to 17β-OH steroids in the ovary or testes. Because the 17β-OH group is essential for binding to the intracellular receptor protein, the addition of a hydrogen atom at C-17 significantly increases biologic potency. For example, the conversion of Δ4-A to T by this enzyme increases androgenicity some sevenfold to 10-fold.
The introduction of a hydrogen atom in the alpha plane at C-5 of T through the action of the 5α—reductase increases the expression of androgenicity in most tissues since the dissociation rate of DHT is much less than that of T from the intracellular androgen receptor. Thus, even though it is present at a lower concentration, DHT will be bound to a greater extent than T.5 It should also be noted that the conversion of T to DHT is essentially unidirectional. Δ4-A and T can also be converted to 5β-androgens, by a 5β-reductase, but this is a less important pathway and probably is active only in splanchnic tissue in mammals but is more widespread and important in avian species.6 It should be noted that the 5β-reduced androgens can also be converted to 3α- or 3β-hydroxy compounds by the action of the appropriate 3-steroid dehydrogenase. Because of the configuration of the 5β-reduced compounds, they bind poorly to the receptor and have minimal activity. Through the action of 3α- or 3β-hydroxy steroid dehydrogenase, DHT is reversibly converted to 5α-androstane, 3α, 17β-diol or 5α-androstane-3β, 17β-diol. The 3α-hydroxysteroid is a member of the aldo-keto reductase superfamily (AKR), of which there are several isoforms.7 3α-diol has significant androgenic activity in certain tissues, but the activity of the 3β-diol is uncertain.8
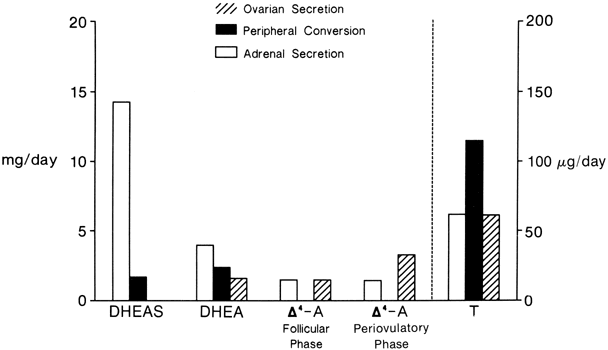
The sources and amounts of the androgens produced daily are shown in Figures 2 and 3. DHEAS is almost exclusively adrenal in origin, but only 50% of DHEA is secreted by the adrenals.1 DHEAS and DHEA plasma levels are age dependent, being low in early childhood and rising between 6 and 8 years of age to plateau in late puberty.9,10
|
|
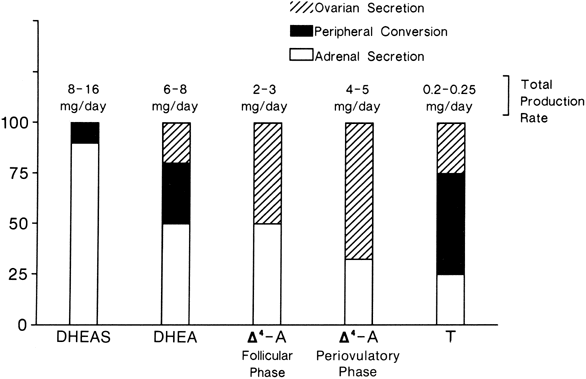
Δ5-A-diol has androgenic activity, but because of its ability to bind to the estrogen receptor, it has estrogenic activities as well. However, other than being a precursor of testosterone, its biologic significance is uncertain. Its production rate has been reported as 1 mg/day in young women.11
Δ4-A is an important androgen in women because of its biopotentiality as a prehormone through its conversion to T or estrone (E)in peripheral tissues. Circulating Δ4-A arises about equally from the adrenal and ovary with only a small amount arising from precursor conversion in nonglandular tissue (see Figs. 2 and 3).1 There is a slight increase in the concentration of Δ4-A in midcycle. Plasma Δ4-A exhibits a diurnal change synchronous with cortisol, although the magnitude of the swing throughout the day is much less than that of cortisol or DHEA because of the significant ovarian source that does not exhibit a diurnal rhythm.12
In normal women, approximately 25% of circulating T is derived from the adrenals and 25% from the ovaries. The other 50% arises from the peripheral conversion of precursors, primarily Δ4-A, with a small amount from DHEA (see Figs. 2 and 3). It should be noted that although the liver contains abundant 17β-hydroxysteroid dehydrogenase, it is a source of minimal amounts of circulating T, for reasons discussed below.
Neither the adrenal nor the ovary secretes DHT, and circulating DHT arises from the peripheral conversion of precursor steroids. In women, the major source of DHT is circulating Δ4-A, but T contributes a small amount.13,14 The liver is also not a source for circulating DHT.15
The adrenal secretion of DHEA and Δ4-A is under the control of adrenocorticotropic hormone (ACTH), but whether ACTH also stimulates the secretion of DHEAS is uncertain.1 Administered as a pulse or an infusion, ACTH causes a threefold rise in DHEA and a twofold to threefold increase in Δ4-A, but the increase in T is smaller and may be because of the peripheral conversion of the increased amounts of precursors. Prolactin receptors have been shown on adrenal cells, and women with hyperprolactinemia may have elevated androgen levels16; but it is not clear whether prolactin plays a role in normal adrenal androgen secretion. The marked increase in the secretion of DHEAS at puberty has raised the question about an additional adrenal androgen-stimulating hormone, since neither ACTH nor cortisol increases then. A pituitary polypeptide, different from ACTH and prolactin, has been suggested as stimulating adrenal androgen secretion,17 but whether such a polypeptide exists remains conjectural.
The ovarian cellular elements that produce androgens include the stroma, theca, and corpus luteum cells.18 A and T are obligatory intermediates in the synthesis of estrone (E) and estradiol (E) by the developing follicles. The theca cells of the follicle stimulated by luteinizing hormone (LH) synthesize Δ4-A and T, and the granulosa cells under follicle-stimulating hormone control aromatize them to estrogens (Fig. 4).18 The corpus luteum cells also secrete androgens as do the stromal cells. Any circumstance that causes dysrhythmia of folliculogenesis may result in excessive ovarian androgen production. In postmenopausal women, the stromal cells are the source of the ovarian androgens.
|

Plasma levels of DHEA, DHEAS, Δ4-A, and T are lower in postmenopausal women.19 The decrease in DHEAS and DHEA is age dependent, but the decrease in Δ4-A and T occurs primarily at or before the menopause and is more marked for Δ4-A than for T.19 The postmenopausal ovary continues to secrete T from stromal cells but is not a major source for Δ4-A.20,21
ANDROGEN TRANSPORT
In the blood, the androgens circulate primarily bound to plasma proteins, and, to a far lesser extent, in the nonprotein-bound, or free, form. The major binding proteins are sex hormone-binding globulin (SHBG) and albumin, although a small percent of the androgens circulate bound to cortisol-binding globulin (CBG).22,23
The steroid structural characteristics that are essential for high-affinity binding to SHBG are a 17β-OH group and a 5α-hydrogen atom. A Δ4 in ring A (double bond in ring A) or an aromatic A ring decreases the binding affinity; for example, Δ4-A with a 17-keto group and a Δ4-unsaturated bond has a relative binding affinity of only 1. 4% compared with 100% for T. E with a 17-OH group, but an aromatic A ring has a relative binding affinity of only 60% of that of T. DHT with both the 17β-OH and the 5α-hydrogen atom has more than a twofold higher binding affinity than T (Table 2).24
Table 2. Equilibrium Association Constants (KA) (M-1) of Steroid Hormone—Protein Complexes in Normal Serum at 37°C
Steroid | Albumin | Globulin |
Dihydrotestosterone | 4×104 | 6×109 |
Testosterone | 4×104 | 2×109 |
Androstenedione | 2×104 | 0.03×109 |
Dehydroepiandrosterone | 4×104 | 0.07×109 |
5α-Androstane-3β-17β-diol | 18×104 | 1.3×109 |
(Data from Dunn JF, Nisula BC, Rodbard D: Transport of steroid hormones: Binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab 53:58, 1981, and Mendel CM: The free hormone hypothesis: A physiologically based mathematical model. Endocr Rev 10:232, 1989).
The distribution of circulating T among the binding proteins is shown in Table 3. In women, the largest proportion (68%) is bound to SHBG, with lesser percentages bound to albumin. There is a small percentage of the steroids bound to CBG.22,23 The binding of T to the proteins obeys the law of mass action; free T is in equilibrium with bound T. The binding capacity of SHBG, therefore, is an important determinant of the percentage of the circulating T that is free or not bound to protein. The circulating level of non-SHBG-bound T may be a better indicator of the biologically active T than either the levels of free or total T.25
Table 3. Distribution as Percent Bound and Free of Steroids in Normal Serum at 37°C}
| Free | Albumin Bound | SHBG Bound* |
Steroid | (%) | (%) | (%) |
DHEA | 4 | 88 | 8 |
Δ4A | 7 | 85 | 8 |
T | 1 | 30 | 69 |
DHT | 1 | 21 | 78 |
3α-A diol | 1 | 71 | 28 |
DHEA, dehydroepiandrosterone; Δ4-A, androstenedione; T, testosterone; DHT, dihydrotestosterone; 3α-A diol, 5α-androstane-3α, 17β-diol.
*Includes a small percentage bound to cortisol-binding globulin for each steroid.
(Data from Dunn JF, Nisula BC, Rodbard D: Transport of steroid hormones: Binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab 53:58, 1981, and Mendel CM: The free hormone hypothesis: A physiologically based mathematical model. Endocr Rev 10:232, 1989).
The idea that the steroid bound to SHBG was inactive, whereas that bound to albumin or free was active, has undergone considerable scrutiny in the past few years.23,26 Certain findings, including immunohistochemical identification of SHBG in cells, the finding of SHBG mRNA in cells other than the liver, the finding of SHBG receptors on cell membranes, and the role of SHBG and androgen acting through the cyclic-AMP pathway27 have all contributed to the re-examination of the role of protein binding of steroids and the entry of steroids into cells for subsequent expression of biologic activity or metabolism.26
The biologic activity of androgens does not necessarily correlate directly with their affinity for SHBG. 28 For example, 3α-diol and 3β-diol have twice the affinity for SHBG compared with T, yet their biologic activity is considerably less than T.
The concentration of SHBG varies with age. In children, the concentration is similar in both sexes but falls during puberty,29 and women have higher concentrations than men during the reproductive years.30 There is a rise with aging in men, the reasons for which are uncertain, but there is little change in postmenopausal women.31 Hyperandrogenic hirsute women usually have a lower-than-normal SHBG concentration. Estrogen therapy, either alone or in the combination-type oral contraceptive, increases the SHBG concentration. The increased endogenous estrogen production of pregnancy doubles the SHBG concentration and total T; free T is reduced from 1% to 0.6%. Thyroid function also influences the SHBG concentration; it is decreased in hypothyroidism and increased in hyperthyroidism. Obesity is associated with a decreased SHBG concentration, and with weight loss, the value returns toward normal.30
Clinical conditions with increased SHBG levels have an increased free E2:T ratio, and those with depressed levels have a decreased circulation-free E2:T ratio. Therefore, one effect of the alterations in SHBG binding dynamics could be to amplify or enhance an estrogenic or androgenic state.32 Other effects of androgen-SHBG binding can be equated with the inverse changes in free androgen that occur with changes in SHBG levels.23
TISSUE METABOLISM AND ACTION
Metabolic Clearance Rate
The rate of metabolism of a steroid by the whole body can be expressed as the volume of blood completely cleared of that steroid per unit time, usually a 24-hour day. This rate is referred to as the metabolic clearance rate (MCR) for that steroid and represents the summation of the clearance rates of the individual organs (e.g., liver, kidney, lung).33,34 The blood production rate of a steroid can be calculated from these data according to the formula:
PB = MCR × PC
where PB is the amount of steroid entering the circulation from all sources and PC is the plasma concentration. It must be emphasized that this method gives only an approximation of the daily production rate, since glandular secretion and MCR can both vary independently during the day.33
The MCRs of the major androgens in healthy women and men are listed in Table 4.11,30,31,32,33,34 The MCR of a steroid is directly correlated with the free fraction of that steroid and, for steroids with a strong affinity to SHBG, inversely correlated with SHBG levels.31,35 Thus, in situations in which SHBG is decreased, as in hyperandrogenism or obesity, the MCRs of T and DHT are increased. In situations in which SHBG is increased, as in hyperthyroidism or estrogen administration, the MCRs of T and DHT are decreased.36,37 The MCRs of Δ4-A and E are also decreased in hypothyroidism, but this is believed because of a decrease in blood flow to splanchnic tissue.38 The MCRs of DHEAS, DHEA, and Δ4-A remain relatively constant despite changes in SHBG.39 However, it should be noted that irrespective of SHBG levels, obesity per se is associated with an increase in MCR for all steroids.40 The MCR of T can also be increased by the induction of metabolizing enzymes in the liver, as seen with medroxyprogesterone administration.41
Table 4. Metabolic Clearance Rates in Liters per Day of the Androgens in Normal Women and Men
| DHEA | DHEAS | DHT | T | Δ4-A |
Women | 1820* | 13.8 | 315 | 485 | 1840 |
Men | 2080 | 15.2 | 650 | 780 | 2390 |
DHEA, dehydroepiandrosterone; DHEAS, dehydroepiandrosterone sulfate; DHT, dihydrotestosterone; T, testosterone; Δ4-A, androstenedione.
*Data from multiple sources.
Androgen Metabolism in the Liver
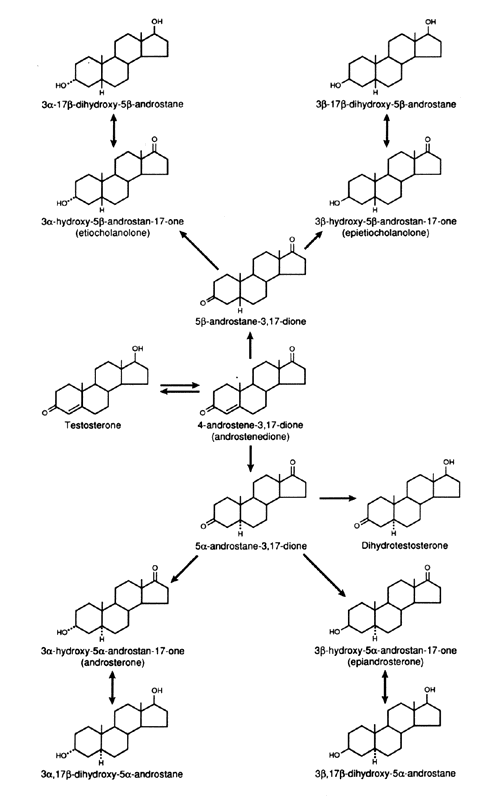
The splanchnic tissues are the major sites of androgen metabolism, and 65%to 90% of the total metabolism occurs in these tissues, of which the liver carries out the major portion. The major sites involved in the metabolism of androgens are the C-3, C-5, and C-17 (Figs. 5 and 6). In the liver, the 5α:5β ratio of reduced androgens can be altered in a number of situations. In hyperthyroidism, the ratio is increased, and in hypothyroidism, it is decreased.42
|
|

The conjugation of androgens to form sulfates or glucuronides occurs primarily at the C-17 and C-3 sites. Because conjugation is rapid, androgens leaving the liver in hepatic vein blood are conjugated primarily as glucuronides, but sulfates are also present. Because there is little cleavage of the glucuronide in tissues, the androgens formed in the liver do not contribute to the blood or tissue pool of unconjugated androgens.15 Androgen glucuronides do not bind to the androgen receptor and are, therefore, biologically inactive. There is some degree of sulfurylation of androgens that occurs in the liver, and the androgen sulfates, especially DHEAS, can be hydrolyzed in other tissues and contribute to the pool of active androgens.
The extraction, or uptake, of androgens by the liver is high and close to 100% for those androgens not bound to SHBG. The binding to SHBG decreases the extraction, and only 40% to 60% of T and 30% to 40% of DHT are extracted by the liver.43,44 However, because of extensive metabolism in the intestinal wall and the liver, the oral administration of T or DHT is an inefficient way to increase overall androgenicity.
Skin and Genital Structures
As noted previously, the free hormone, which is inversely related to the SHBG concentration, is available for entering the cells of the target tissues.23 Recently, T was considered to be the male hormone. However, with advancement of the knowledge of cellular biology, particularly the intracellular events associated with biologic responses to androgen, it was evident that in many androgen-responsive tissues, DHT rather than T mediated the intracellular molecular events associated with responses to androgens.8 As noted previously, because of different binding affinities, DHT is a stronger androgen than is T.5 In tissues with relatively high levels of 5α-reductase, such as the hair follicle and sebaceous gland, the amount of DHT formed from precursors is relatively large and androgenic events are mediated by DHT rather than T. Thus, DHT is responsible for the male differentiation of the prostate and external genitalia, but virilization of the wolffian ducts is due to T.5,45 The levels of 5α-reductase are low in muscle;therefore, muscle development is under T control.8
The concept that steroids bind to and result in the nuclear translocation of a cytoplasmic receptor has altered in the past few years.464748 It has been shown that estrogen receptors exist almost entirely in the nucleus, irrespective of steroid binding, and that estradiol diffuses through the cell membranes and cytoplasm to bind to the receptor in the nucleus. It would appear that the same concept applies to the androgens.48 The receptor is present in the nucleus but not bound to DNA. When androgen binds to its receptor, the latter can then bind to DNA and initiate transcription.46,48
In tissues with high levels of 5α-reductase, when a precursor, DHEAS, DHEA, Δ4-A, or T enters the cell, conversion to DHT occurs through the pathways shown in Figure 1. DHT is the androgen that is primarily responsible for stimulating the androgenic activity. In tissues that have minimal 5α-reductase activity, the same precursors are converted only to T, and it is that steroid that stimulates the androgenic activity.49,50 In either case, the degree of androgenic expression is dependent on the amount of active steroid or precursor steroid in the circulation, the blood flow to the tissue, the extraction of steroid by the tissue, and the enzyme activities of the tissue.
In vitro studies have documented that human skin has the capacity to metabolize androgens and is an androgen-responsive tissue.8 The skin from the genital area of the fetus at 12 to 22 weeks of age has a greater capacity than nongenital skin to convert T to DHT, and the external genital skin of male and female fetuses can transform T to DHT. Thus, 5α-reductase activity in the urogenital tubercle, fold, and swelling is acquired very early in embryogenesis in both male and female fetuses and is not, itself, androgen dependent.45 The production of T by the testes of the male fetus and the in situ conversion to DHT stimulate these structures to form the external male genitalia. In the absence of T production or conversion to DHT, female external genitalia develop. The 5α-reductase activity persists after birth in those structures derived from the urogenital tubercle, fold, and swelling in both sexes. The clitoris is, therefore, androgen responsive.
Valuable knowledge concerning the role of androgen metabolism in sexual differentiation of the internal and external genitalia has been acquired from studies of the skin and genital structures in male pseudohermaphroditism.51,52 In testicular feminization (i.e., complete male pseudohermaphroditism), the intracellular androgen receptor is absent but Müllerian inhibiting factor is fully active. Consequently, both male internal and external genitalia fail to develop, male secondary sex characteristics are not acquired, and the phenotype is female. In the type 2 incomplete form of male pseudohermaphroditism, an autosomal-recessive disorder, there is deficiency of 5α-reductase activity, resulting in deficiency of DHT formation, despite normal T and Δ4-A production. As a consequence of the inadequate DHT formation, there is incomplete differentiation of the male external genitalia, although the internal male genitalia develop normally.53 These findings indicate that 5α-reductase activity and DHT are required for differentiation of the male external genitalia but not the male internal sexual structures. The increased secretion of T at puberty stimulates enlargement of the phallus and induces the male secondary sex characteristics, including an increase in muscle mass, but beard growth and acne are minimal, indicating that these events are DHT dependent.
One of the most interesting aspects of androgen metabolism concerns the growth of body hair.54 Not all hair growth is androgen dependent or androgen responsive. Androgen-dependent hair, or sexual hair, is located on the face, chest, and abdomen (center of the body). Ambisexual hair is stimulated by low levels of androgens and includes axillary hair and the hair on the lower pubic triangle. Nonsexual hair (e.g., eyebrows) is independent of androgen effects.
The control of 5α-reductase activity is different in the external genitalia and skin containing sexual hair.54 The ability of the skin containing sexual hair to form DHT from T increases during puberty as T production increases. Thus, 5α-reductase activity in these areas is induced by T.54 Male secondary sex characteristics (loss of hair, increased sebaceous gland activity) are induced by T during puberty and are dependent on acquisition of 5α-reductase activity necessary for the conversion of T to DHT . There is a direct relationship between the extent of hair growth in the skin of sexual areas and the amount of 5α-reductase activity.54
Many hirsute women have increased production of androgens (DHEA, A, T, or all three), and these are transformed to DHT, especially in hair follicles and sebaceous glands.54 The amount of DHT formed will be increased primarily because of increased precursor production. There is, however, an increase in 5α-reductase activity in most hirsute women.54 In certain instances of hirsutism and male pattern baldness, there is increased formation of DHT from precursors in the hair roots,51,52,54 even though the circulating precursors DHEAS, DHEA, A, and T production are normal in these women. It is believed that an increased 5α-reductase activity is the cause of the hirsutism since the increased 5α-reductase activity would result in increased DHT production from normal amounts of precursors within the hair follicle. Then, after stimulating hair growth, the DHT is metabolized to androstanediol that is conjugated before leaving the hair follicle as 5α-androstanediol-3α, 17β-diol glucuronide(3α-Adiol gluc).54 The latter steroid is believed to be a marker for the increased 5α-reductase activity in these women.51,54,55
The sebaceous glands are also androgen-responsive organs and have the capability of converting precursors to DHT. Sebaceous gland secretory activity is increased in hyperandrogenic women, and seborrhea usually accompanies hirsutism. There is hypertrophy of the sebaceous glands in the skin with acne and an increased 5α-reductase activity.56
There is poor correlation between the plasma level of T, or other androgens, and the severity of hirsutism or acne. However, plasma-free T correlates better than total T with the degree of androgenization,57 and it now appears that many hirsute women have increased androgen production.58 In such women, there is good correlation between the production rate of T and the degree of hirsutism or virilism. As noted, many women with hirsutism but normal androgen production rates have increased 5α-reductase activity and increased circulating levels of 5α-Adiol gluc.54 It has also been suggested that other conjugated androgens, specifically androsterone sulfate and androsterone glucuronide and 3α-diol sulfate, may reflect peripheral androgen activity.59
One of the major causes of hyperandrogenism in women is polycystic ovarian syndrome (PCOS).60,61 There may be overproduction of DHEA, A, T, or all three in this disorder, and the cellular origin of the excess androgens includes the stromal cells, those cells lining atretic follicles, and luteinized stromal cells. Most women with PCOS have elevated LH levels,62 although this may not be obvious in a single blood sample. A major component of PCOS is insulin resistance,54,63,64 which leads to glucose intolerance and diabetes mellitus.65
Androgen-secreting tumors of the ovary include thecomas, luteomas, hilar cell tumors, and arrhenoblastomas.66 Androgen-secreting ovarian tumors may be gonadotropin dependent and responsive and occur with increased frequency in the LH-stimulated polycystic ovary. T levels are usually markedly elevated (greater than 2 ng/mL; greater than 7 nmol/L) in association with ovarian tumors but only occasionally reach such levels in PCOS.67
Adrenal disorders causing excessive androgen production include the various forms of congenital adrenal hyperplasia (CAH), the most common of which is that associated with 21-hydroxylase deficiency.68 This can present in a variable manner from abnormal genitalia and adrenal insufficiency at or soon after birth to postpubertal hirsutism.68 The less-common forms of CAH are those associated with deficient 11β-hydroxylase69 and 3β-hydroxy-Δ5— steroid dehydrogenase deficiency.70 In the former, the deficiency of 11β-hydroxylase activity can cause hypertension and signs of androgen excess and is associated with an elevation of 11-deoxycortisol.69 In subjects with a deficiency of the 3β-hydroxy-Δ5— steroid dehydrogenase enzyme system that converts 3β-hydroxy-Δ5— steroids to 3-keto-Δ4-steroids, there is an increase in the secretion of 17α-hydroxy pregnenolone and DHEA and a relative decrease in the secretion of progesterone and Δ4-A.70 The increased DHEA results in signs of hyperandrogenism, presumably because the enzymes in the peripheral tissues are not defective to the same degree as in the adrenals and ovaries. The signs of hyperandrogenism depend on the degree of enzyme deficiency. The classic type presents at or near birth, usually with abnormal external genitalia and severe symptoms of adrenal insufficiency. The nonclassic type presents at late childhood or adolescence with varying signs and symptoms of hyperandrogenism. The diagnosis depends on finding an increase in the levels of 17α-hydroxypregnenolone or DHEA.71 The frequency with which this syndrome is seen is variable depending on the clinic population.
The nonclassic 21-hydroxylase deficiency is transmitted as an autosomal-recessive trait and becomes apparent in the affected person in later childhood or adolescence.68 The 21-hydroxylase deficiency gene is located on chromosome 6, and there appears to be an increased frequency of the HLAB14,DR1 haplotype. In Ashkenazi Jews, the frequency of the disorder has been reported to be as high as one in 30, but in mixed non-Jewish whites, the frequency is much less(1:1000).68 The incidence in clinical series is, thus, dependent on the population base of that clinic. Affected persons have an increased level of 17α-OH progesterone, but this is often demonstrable only after ACTH stimulation.72 Interestingly, there is a cryptic form of the disorder in which the person has the genetic and biochemical defect but is asymptomatic.
Benign or malignant tumors of the adrenals may also be a source of excess androgens. The functioning adrenal tumors usually have relative deficiency of 3β-hydroxy-Δ5— steroid dehydrogenase activity; and, consequently, most produce primarily Δ5— steroids, particularly DHEA and DHEAS. However, a few T-secreting adrenal tumors have been reported.
As noted earlier, the population base of clinics is variable so that reports on the most frequent cause of hyperandrogenism (adrenal, ovarian, or idiopathic) also are variable. However, PCOS is believed by many to be the most common cause. This is confounded because androgen excess itself can result in PCOS, and thus a combined adrenal ovarian defect can be demonstrable.73
Adipose Tissue
The capability of adipose tissue to metabolize androgens is well recognized. Foremost among the androgen metabolic activities of adipose tissue is its ability to aromatize androgens to estrogens,74,75 and obese men and postmenopausal women can have elevated levels of estrogens. Although adipose tissue does have the 17β-hydroxysteroid dehydrogenase enzyme system, the interconversion of Δ4-A and T is not increased in obesity as is the aromatization.76
In obese persons, the location of the adiposity appears to play a role in their androgen metabolism.77 In women with most of the adipose tissue in the abdominal area, there is a tendency for more signs of hyperandrogenism than in women with the obesity in the hip and buttocks area.78 The amount of aromatization also differs depending on the waist-hip ratio. Those women with high ratios have lower aromatization.78
As noted previously, the production rates of androgens are usually increased in obese women, but this is primarily because of an increase in the adrenal secretion of precursors. The conversion rate of A to E1 increases with age and is the major source of estrogen in postmenopausal women.75,79,80 The excessive production of E1 in the fat tissue of postmenopausal obese women can result in abnormal uterine bleeding. This pathologic state may also result from excessive A production with a normal A-to-E1 conversion rate. The physiologic significance of other known aspects of androgen metabolism in fat tissue remains largely speculative.
Brain
Masculinization of the brain in males is induced by androgens early in fetal life.8,49 Female animals exhibit male psychic and sexual activity after an injection of T during a critical neonatal period. Data indicate that this effect is motivated by way of the estrogen produced from the peripheral conversion of T. Finally, 5α-reductase activity is present in the hypothalamus and the anterior pituitary gland. Although in men androgens play a role in the control of gonadotropin secretion, in women the estrogens are the steroids that are most important. Aromatization occurs in the hypothalamus, but in healthy women, most of the estrogen controlling gonadotropin release comes from the circulation.
REFERENCES
Longcope C: Adrenal and gonadal androgen secretion in normal females. Clin Endocrinol Metab 15: 213– 228, 1986 |
|
King RJB, Mainwaring WIP: Steroid-Cell Interactions. Baltimore, MD:University Park Press, 1974 |
|
Penning TM: Molecular endocrinology of hydroxysteroid dehydrogenases. Endocr Rev 18: 281– 305, 1997 |
|
Labrie F, Luu-The V, Lin S-X et al: The key role of 17B-hydroxysteroid dehyrogenases in sex steroid biology. Steroids 62: 148– 158, 1997 |
|
Grino PB, Griffin JE, Wilson JD: Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endo 126: 1165– 1172, 1990 |
|
Mauvais-Jarvis P, Kuttenn F, Mowszowicz I: Androgen metabolism in human skin: Importance of dihydrotestosterone formation in normal and abnormal target cells. In Molinatti G, Martini L, James VHT (eds): Androgenization in women, pp 47–63. New York, Raven Press, 1983 |
|
Penning TM, Burczynski ME, Jez JM et al: Human 3a-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J 351: 67– 77, 2000 |
|
Mooradian AD, Morley JE, Korenman SG: Biological actions of androgens. Endocrine Reviews 8: 1– 28, 1987 |
|
Ducharme J-R, Forest MG, De Peretti E et al: Plasma adrenal and gonadal sex steroids in human pubertal development. J Clin Endocrinol Metab 42: 468– 476, 1976 |
|
De Peretti E, Forest MG: Unconjugated dehydroepiandrosterone plasma levels in normal subjects from birth to adolescence in human: The use of a sensitive radioimmunoassay. J Clin Endocrinol Metab 43: 982– 991, 1976 |
|
Bird CE, Morrow L, Fukomoto Y et al: 5 -Androstenediol: Kinetics of metabolism and binding to plasma proteins in normal men and women. J Clin Endocrinol Metab 43: 1317– 1322, 1976 |
|
Parker LN: Adrenal Androgens in Clinical Medicine. New~York, Academic Press, Inc, 1989 |
|
Ito T, Horton R: Source of plasma dihydrotestosterone in man. J Clin Invest 50: 1621– 1627, 1971 |
|
Mahoudeau JA, Bardin CW, Lipsett MB: The metabolic clearance rate and origin of plasma dihydrotestosterone in man and its conversion to the 5α-androstanediols. J Clin Invest 50: 1338– 1344, 1971 |
|
Horton R, Lobo R: Peripheral androgens and the role of androstanediol glucuronide. J Clin Endocrinol Metab 15: 293– 306, 1986 |
|
Jones DL, Jacobs HS, James VHT: The relationship between plasma prolactin and dehydroepiandrosterone and dehydroepiandrosterone sulphate levels in patients with hyperprolactinemia. In Genazzani AR, Thijssen JHH, Siiteri PK (eds): Adrenal Androgens, pp 83–87. New York, Raven Press, 1980 |
|
Odell W, Parker L: Control of adrenal androgen secretion. In Genazzani AR (ed): Adrenal Androgens, p 27. New York, Raven Press, 1980 |
|
Yen SSC: The Human Menstrual Cycle. In Yen SSC, Jaffee RB (eds):Reproductive Endocrinology: Physiology, Pathophysiology and Clinical Management, pp 200–236. Philadelphia, W.B. Saunders Co, 1986 |
|
Longcope C, Franz C, Morello C et al: Steroid and gonadotropin levels in women during the peri-menopausal years. Maturitas 8: 189– 196, 1986 |
|
Judd HL, Fournet N: Changes of ovarian hormonal function with aging. Exp Gerontol 29: 285– 298, 1994 |
|
Longcope C, Hunter R, Franz C: Steroid secretion by the postmenopausal ovary. Am J Obstet Gynecol 138: 564– 568, 1980 |
|
Dunn JF, Nisula BC, Rodbard D: Transport of steroid hormones: Binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab 53: 58– 68, 1981 |
|
Mendel CM: The free hormone hypothesis: A physiologically based mathematical model. Endocr Rev 10: 232– 274, 1989 |
|
Westphal U: Steroid-Protein Interactions II. Monographs in Endocrinology, pp 250–257. New York, Springer-Verlag, 1986 |
|
van den Beld AW, de Jong FH, Grobbee DE et al: Measures of bioavailable serum testosterone and estradiol and their relationships with muscle strength, bone density, and body composition in elderly men. JCEM 85: 3276– 3282, 2000 |
|
Joseph DR: Structure, function, and regulation of androgen-binding protein/sex hormone-binding globulin. Vitamins and Hormones, pp 197–280. Academic Press, Inc, 1994 |
|
Rosner W, Hryb DJ, Khan MS et al: Androgen and estrogen signaling at the cell membrane via G-proteins and cyclic adenosine monophosphate. Steroids 64: 100– 106, 1999 |
|
Siiteri PK, Simberg NH: Changing concepts of active androgens in blood. J Clin Endocrinol Metab 15: 247– 258, 1986 |
|
Belgorosky A, Rivarola MA: Progressive increase in nonsex hormone-binding globulin-bound testosterone and estradiol from infancy to late puberty in girls. J Clin Endocrinol Metab 67: 234– 237, 1988 |
|
Vermeulen A: Physiology of the testosterone-binding globulin in man. Ann N Y Acad Sci 538: 103– 111, 1988 |
|
Longcope C, Hui SL, Johnston CC, Jr: Free estradiol, free testosterone and sex hormone binding globulin in peri-menopausal women. J Clin Endocrinol Metab 64: 513– 518, 1987 |
|
Rosner W: The functions of corticosteroid-binding globulin and sex hormone-binding globulin: Recent advances. Endocr Rev 11: 80– 91, 1990 |
|
Baird DT, Horton R, Longcope C et al: Steroid dynamics under steady-state conditions. Recent Prog Horm Res 25: 611– 664, 1969 |
|
Gurpide E: Experimental designs used to estimate rates of steroid production and metabolism in vivo and in vitro. Ann N Y Acad Sci 595: 165– 172, 1990 |
|
Siiteri PK, Murai JT, Hammond GL et al: The serum transport of steroid hormones. Recent Prog Horm Res 38: 457– 510, 1982 |
|
Mattheij JA, Swarts JJ, Lokerse P et al: Effect of hypothyroidism on the pituitary-gonadal axis in the adult female rat. J Endocrinol 146: 87– 94, 1995 |
|
Ridgway EC, Maloof F, Longcope C: Androgen and oestrogen dynamics in hyperthyroidism. J Endocrinol 95: 105– 115, 1982 |
|
Pirke KM, Doerr P: Age related changes and interrelationships between plasma testosterone, oestradiol and testosterone-binding globulin in normal adult males. Acta Endocrinol 74: 792– 800, 1973 |
|
Kurtz BR, Givens JR, Komindr S et al: Maintenance of normal circulating levels of 4 -androstenedione and dehydroepiandrosterone in simple obesity despite increased metabolic clearance rates: Evidence for a servo-control mechanism. J Clin Endocrinol Metab 64: 1261– 1267, 1987 |
|
Kirschner MA, Samojlik E, Silber D: A comparison of androgen production and clearance in hirsute and obese women. J Steroid Biochem 19: 607– 614, 1983 |
|
Gordon GG, Southern AL, Tochimoto S et al: Effect of medroxyprogesterone acetate (Provera) on the metabolism and biological activity of testosterone. J Clin Endocrinol Metab 30: 449, 1970 |
|
Gallagher TF, Fukushima DK, Noguchi S et al: Recent studies in steroid hormone metabolism in man. Recent Prog Horm Res 22: 283– 303, 1966 |
|
Longcope C, Sato K, McKay C et al: Aromatization by splanchnic tissue in men. J Clin Endocrinol Metab 58: 1089– 1093, 1984 |
|
Ishimaru T, Edmiston WA, Pages L et al: Splanchnic extraction and conversion of testosterone and dihydrotestosterone in man. J Clin Endocrinol Metab 46: 528– 533, 1978 |
|
Saenger P: Abnormal sex differentiation. J Pediatr 104: 1– 17, 1984 |
|
Gorski J, Welshons WV, Sakai D et al: Evolution of a model of estrogen action. Recent Prog Horm Res 42: 297– 329, 1986 |
|
Rories C, Spelsberg TC: Ovarian steroid action on gene expression:Mechanisms and models. Annu Rev Physiol 51: 653– 681, 1989 |
|
Carson-Jurica MA, Schrader WT, O'Malley BW: Steroid receptor family:Structure and functions. Endocr Rev 11: 201– 220, 1990 |
|
Martini L, Celotti F, Lechuga MJ et al: Androgen meta-bolism in different target tissues. Ann N Y Acad Sci 595: 184– 198, 1990 |
|
Wilson JD: The role of androgens in male gender role behavior. Endocr Rev 20: 726– 737, 1999 |
|
Horton R, Hawks D, Lobo R: 3α,17β-androstanediol glucuronide in plasma:a marker of androgen action in idiopathic hirsutism. J Clin Invest 69: 1203– 1206, 1982 |
|
Kirschner MA, Samojlik E, Szmal E: Clinical usefulness of plasma androstanediol glucuronide measurements in women with idiopathic hirsutism. J Clin Endocrinol Metab 65: 597– 601, 1987 |
|
Imperato-McGinley J: Sexual differentiation: Normal and abnormal. In Martini L, James VHT (eds): Fetal Endocrinology and Metabolism, pp 231–307. New York, Academic Press, 1983 |
|
Azziz R, Carmina E, Sawaya ME: Idiopathic Hirsutism. Endocr Rev 21: 347– 362, 2000 |
|
Thiboutot D: Normal physiology of the pilosebaceous unit. In Azziz R, Nestler JE, Dewailly D (eds): Androgen Excess Disorders in Women, pp 103–114. Philadelphia, PA, Lippincott-Raven, 1997 |
|
Mauvais-Jarvis P: Regulation of androgen receptor and 5α-reductase in the skin of normal and hirsute women. J Clin Endocrinol Metab 15: 307– 317, 1986 |
|
Ehrmann DA, Rosenfield RL: An endocrinological approach to the patient with hirsutism. J Clin Endocrinol Metab 71: 1– 4, 1990 |
|
Biffignandi P, Massucchetti C, Molinatti GM: Female hirsutism:Pathophysiological considerations and therapeutic implications. Endocr Rev 5: 498– 513, 1984 |
|
Matteri RK, Stanczyk FZ, Gentzschein EE et al: Androgen sulfate and glucuronide conjugates in nonhirsute and hirsute women with polycystic ovarian syndrome. Am J Obstet Gynecol 161: 1704– 1709, 1989 |
|
Carmina E, Lobo R: Do hyperandrogenic women with normal menses have polycystic ovary syndrome? Fertil Steril 71: 319– 322, 1999 |
|
Kahsar-Miller M, Nixon C, Boots LR et al: Prevalence of polycystic ovary syndrome (PCOS) in first-degree relatives of patients with PCOS. Fertil Steril 75: 53– 58, 2001 |
|
Kalro BN, Loucks TL, Berga SL: Neuromodulation in polycystic ovary syndrome. Obstet Gynecol Clin North Am 28: 35– 62, 2001 |
|
Venkatesan AM, Dunaif A, Corbould A: Insulin resistance in polycystic ovary syndrome: Progress and paradoxes. Recent Prog Horm Res 56: 295– 308, 2001 |
|
Zacur HA: Polycystic ovary syndrome, hyperandrogenism, and insulin resistance. Obstet Gynecol Clin North Am 28: 21– 33, 2001 |
|
Legro RS: Diabetes prevalence and risk factors in polycystic ovary syndrome. Obstet Gynecol Clin North Am 28: 99– 109, 2001 |
|
Givens JR: Hirsutism and hyperandrogenism. Adv Intern Med 21: 221, 1976 |
|
Rittmaster RS, Loriaux DL: Hirsutism. Ann Intern Med 106: 95– 107, 1987 |
|
White PC, Speiser PW: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 21: 245– 291, 2000 |
|
White PC: Steroid 11 beta-hydroxylase deficiency and related disorders. Endocrinol Metab Clin North Am 30: 61– 79, 2001 |
|
Pang S: Congenital adrenal hyperplasia owing to a 3 beta-hydroxysteroid dehydrogenase deficiency. Endocrinol Metab Clin North Am 30: 81– 99, 2001 |
|
Lobo R, Goebelsmann U: Evidence for reduced 3β-ol-hydroxysteroid dehydrogenase activity in some women thought to have polycystic ovary syndrome. J Clin Endocrinol Metab 53: 394– 400, 1981 |
|
Azziz R, Zacur HA: 21-hydroxylase deficiency in female hyperandrogenism:Screening and diagnosis. J Clin Endocrinol Metab 69: 577– 584, 1989 |
|
Moran C, Azziz R: The role of the adrenal cortex in polycystic ovary syndrome. Obstet Gynecol Clin North Am 28: 63– 75, 2001 |
|
Toscano V: Dihydrotestosterone metabolism. J Clin Endocrinol Metab 15: 279– 292, 1986 |
|
Longcope C: Methods and results of aromatization studies in vivo. Cancer Res 42: 3307s– 3311s, 1982 |
|
Longcope C, Baker R, Johnston CC, Jr: Androgen and estrogen metabolism in relationship to obesity. Metabolism 35: 235– 237, 1986 |
|
Killinger DW, Perel E, Daniilescu D et al: Influence of adipose tissue distribution on the biological activity of androgens. Ann N Y Acad Sci 595: 199– 211, 1990 |
|
Kirschner MA, Samojlik E, Drejka M et al: Androgen-estrogen metabolism in women with upper body versus lower body obesity. J Clin Endocrinol Metab 70: 473– 479, 1990 |
|
Longcope C: Hormone dynamics at the menopause. Ann N Y Acad Sci 592: 21– 30, 1990 |
|
Bulun SE, Simpson ER: Competitive reverse transcription-polymerase chain reaction analysis indicates that levels of aromatase cytochrome P450 transcripts in adipose tissue of buttocks, thighs, and abdomen of women increase with advancing age. JCEM 78:428– 432, 1994 |